


近日,学院研究团队在电催化生物质高值转化方面取得重要进展,研究成果以“In Situ Generated Dual Oxyanions Enable Bifunctional Synergy to Overcome Dehydrogenation Limitations in HMF Electrooxidation”(原位生成的双重含氧阴离子通过双功能协同突破HMF电氧化中的脱氢限制)为题发表在国际知名期刊《Chemical Science》(中科院化学一区,IF= 8.4,TOP期刊)上。
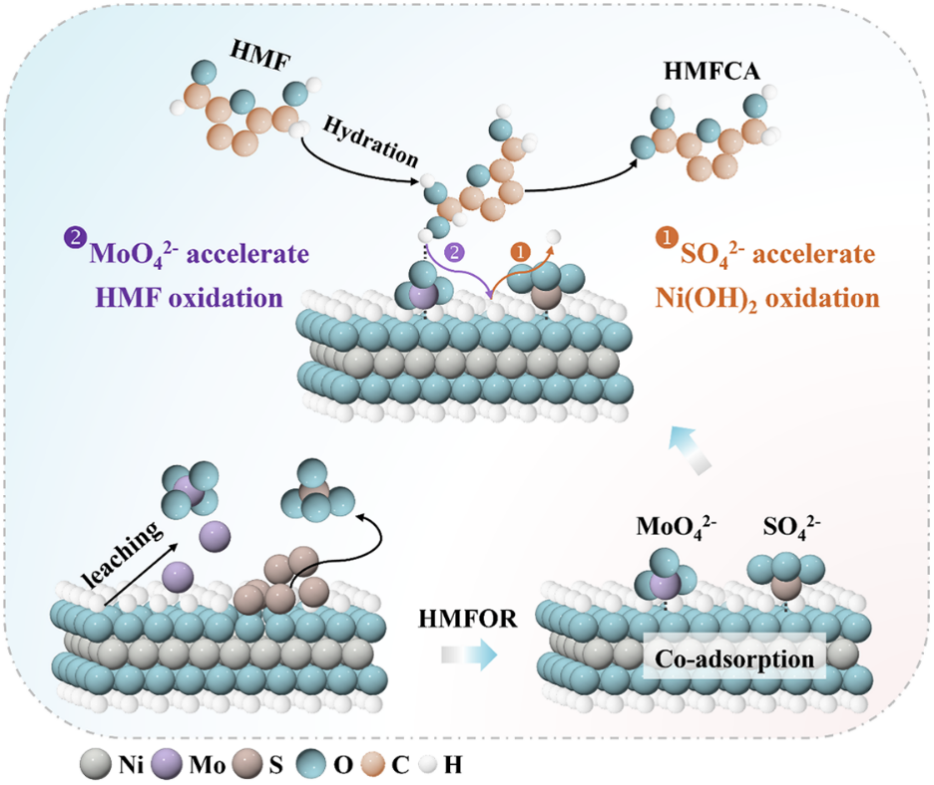
图1双重含氧阴离子对HMFOR中两步脱氢的协同调控示意图
5-羟甲基糠醛(HMF)作为重要的“生物质平台分子”,可电氧化生成2,5-呋喃二甲酸(FDCA)。该产物是合成可降解聚合物聚呋喃二甲酸乙二醇酯(PEF)的关键单体,能够替代化石基聚对苯二甲酸乙二醇酯(PET),对缓解“白色污染”与降低化工产业碳排放意义重大。电化学HMF氧化反应(HMFOR)可在常温常压下进行,成为当前研究热点。然而,HMFOR的工业化长期受限于高效电催化剂的缺乏。镍基材料虽然成本低、活性好,但其性能受到两个脱氢步骤的制约:一是Ni(OH)2向活性Ni3+物种转化的动力学缓慢,二是HMF分子在催化剂表面的吸附与脱氢效率不足。传统策略往往顾此失彼,且过度加速Ni(OH)2脱氢易导致催化剂不可逆氧化失活,从而形成了活性与稳定性难两全的困境。
基于此,研究团队提出了一种双氧阴离子协同吸附策略,通过构建Mo掺杂的Ni3S2修饰Ni(OH)x电催化剂(Mo-Ni3S2/Ni(OH)x),在电化学反应过程中原位生成SO42-和MoO42-两种含氧阴离子,并实现它们在Ni(OH)x表面的共吸附。结果表明,Mo-Ni3S2/Ni(OH)x展现出优异的HMFOR性能:仅需1.46 V vs. RHE即可达到100 mA cm-2的电流密度,是仅吸附SO42-的Ni3S2/Ni(OH)x的2.5倍。经过8次循环测试后,保持100%HMF转化率、98%FDCA选择性和94%法拉第效率。在膜电极电解池(MEA)中,该催化剂在1.9 V下经过20次循环仍保持近100%的FDCA产率,展现出良好的实际应用潜力。结合原位拉曼光谱与密度泛函理论计算,团队首次阐明了两种结构相似的氧阴离子在共吸附体系中的差异化协同调控机制:SO42-主要与Ni(OH)2表面的氢原子形成氢键,将Ni(OH)2脱氢生成Ni3+的能垒从2.50 eV降低至2.26 eV,加速了活性Ni3+物种的形成;MoO42-则优先与HMF分子的氢原子相互作用,促进HMF的吸附及其脱氢动力学。这种“双功能协同”效应巧妙地将两个脱氢步骤解耦并分别加速,在不牺牲稳定性的前提下大幅提升了反应速率。
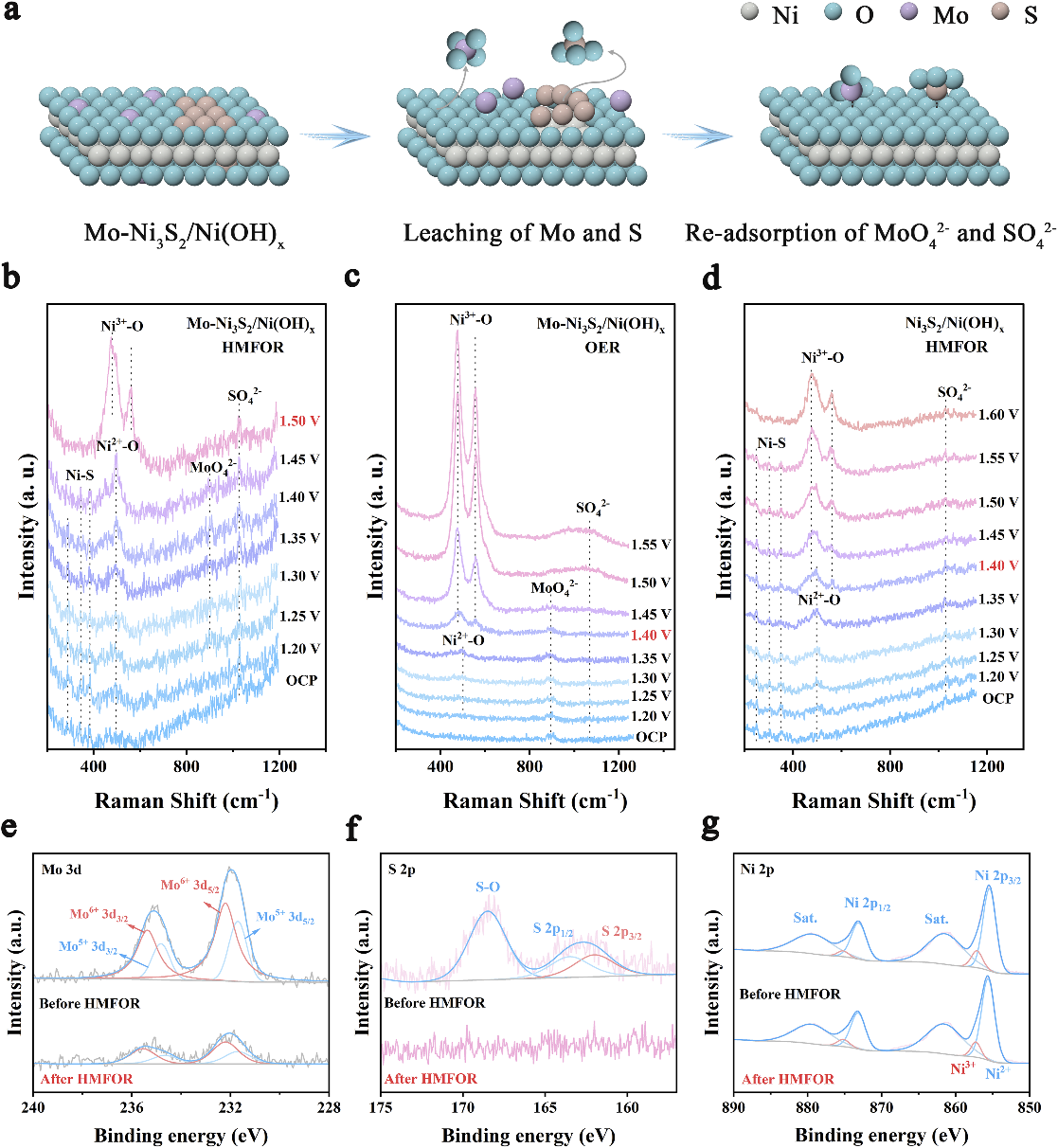
图2Mo-Ni3S2/Ni(OH)x的重构机制
研究还揭示了一个重要现象:原位自生成的含氧阴离子比外加的氧阴离子具有更显著的促进效果。这是由于外加氧阴离子会与HMF和OH-在活性位点上发生竞争吸附,反而抑制了HMF的吸附,而原位生成的氧阴离子能够有效地优化HMF的结合强度。该工作首次阐明了类似含氧阴离子在复杂共吸附体系中对HMF氧化的差异化调控机制,为生物质高值化利用领域高效电催化剂的设计提供了全新的理论指导。
论文通讯作者为学院包福喜副教授和于锋教授;第一作者为学院2023级博士研究生肖悦,共同作者包括连梦如、刘嘉妮以及李雪雪。
(图文:包福喜;初审:张海洋;复审:刘平;终审:徐炜杰)